









Contenido
Esclerosis tuberosa de Bourneville
Qué es ?
La esclerosis tuberosa de Bourneville es una enfermedad genética compleja caracterizada por el desarrollo de un tumor benigno (no canceroso) en diferentes partes del cuerpo. Luego, estos tumores pueden ubicarse en la piel, el cerebro, los riñones y otros órganos y tejidos. Esta patología también puede ocasionar serios problemas en el desarrollo del individuo. Sin embargo, las manifestaciones clínicas y la gravedad de la enfermedad varían de un paciente a otro.
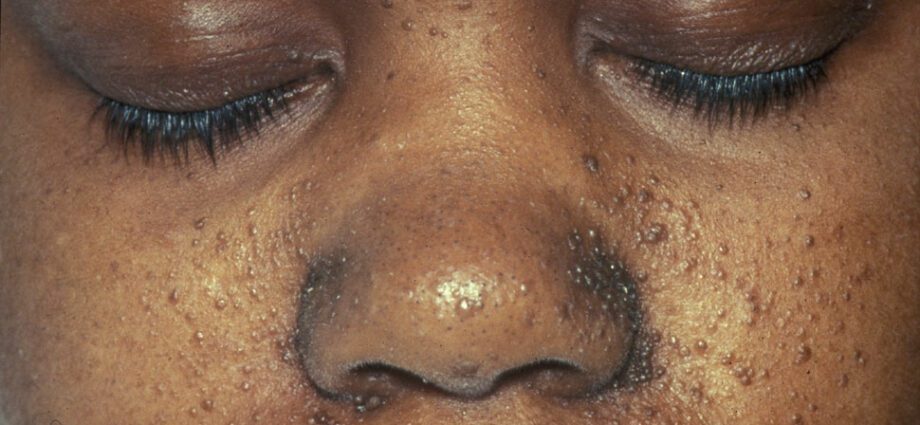
Las anomalías cutáneas asociadas son generalmente similares a manchas en la piel o áreas donde la piel es más clara que en el resto del cuerpo. El desarrollo de tumores en la cara se llama angiofibroma.
En el contexto del daño cerebral, los signos clínicos son ataques epilépticos, problemas de comportamiento (hiperactividad, agresividad, discapacidad intelectual, problemas de aprendizaje, etc.). Algunos niños con la enfermedad incluso tienen alguna forma de autismo, trastornos del desarrollo, que afectan las interacciones sociales y la comunicación. Los tumores cerebrales benignos también pueden causar complicaciones que pueden ser fatales para el sujeto.
El desarrollo de tumores en los riñones es común en personas con esclerosis tuberosa. Esto puede causar graves complicaciones en la función renal. Además, pueden desarrollarse tumores en el corazón, los pulmones y la retina. (2)
Es una enfermedad rara, cuya prevalencia (número de casos en una población determinada en un momento determinado) asciende a 1/8 a 000/1 personas. (15)
Síntomas
Las manifestaciones clínicas asociadas a la esclerosis tuberosa de Bourneville varían según los órganos afectados. Además, los síntomas asociados con la enfermedad varían mucho de un individuo a otro. Con síntomas que van de leves a graves.
Los síntomas más ampliamente identificados de esta enfermedad incluyen ataques epilépticos, trastornos cognitivos y del comportamiento, anomalías cutáneas, etc. Los órganos más afectados son: el cerebro, el corazón, los riñones, los pulmones y la piel.
El desarrollo de tumores malignos (cancerosos) es posible en esta enfermedad, pero son raros y afectan principalmente a los riñones.
Los signos clínicos de la enfermedad en el cerebro se originan en ataques a diferentes niveles:
- daño a los tubérculos corticales;
- nódulos ependimarios (NEE);
- astrocitomas ependimarios gigantes.
Resultan en: desarrollo de retraso mental, dificultades de aprendizaje, trastornos del comportamiento, agresividad, trastornos de atención, hiperactividad, trastornos obsesivo-compulsivos, etc.
El daño renal se caracteriza por el desarrollo de quistes o angiomiolipomas. Estos pueden provocar dolor de riñón e incluso insuficiencia renal. Si se nota un sangrado abundante, puede deberse a una anemia grave o presión arterial alta. También pueden ser visibles otras consecuencias más graves pero raras, en particular el desarrollo de carcinomas (tumor de las células constituyentes del epitelio).
El daño ocular puede ser similar a las manchas visibles en la retina, provocando alteraciones visuales o incluso ceguera.
Las anomalías cutáneas son numerosas:
- máculas hipomelanicas: que provocan la aparición de manchas claras en la piel, en cualquier parte del cuerpo, como consecuencia de una deficiencia de melanina, una proteína que da color a la piel;
- la aparición de manchas rojas en la cara;
- parches descoloridos en la frente;
- otras anomalías cutáneas, que dependen de un individuo a otro.
Las lesiones pulmonares están presentes en 1/3 de los pacientes con un ligero predominio femenino. Los síntomas asociados son entonces dificultades respiratorias más o menos graves.
Los orígenes de la enfermedad
El origen de la enfermedad es genético y hereditario.
La transmisión implica mutaciones en los genes TSC1 y TSC2. Estos genes de interés entran en juego en la formación de proteínas: hamartina y tuberina. Estas dos proteínas permiten, mediante un juego interactivo, regular la proliferación celular.
Los pacientes con la enfermedad nacen con al menos una copia mutada de estos genes en cada una de sus células. Estas mutaciones luego limitan la formación de hamartina o tubertina.
En el contexto en el que las dos copias del gen están mutadas, impiden por completo la producción de estas dos proteínas. Por tanto, esta deficiencia proteica ya no permite que el organismo regule el crecimiento de determinadas células y, en este sentido, conduce al desarrollo de células tumorales en diferentes tejidos y / u órganos.
Los factores de riesgo
Los factores de riesgo para desarrollar tal patología son genéticos.
De hecho, la transmisión de la enfermedad es eficaz a través de un modo autosómico dominante. O bien, el gen mutado de interés se encuentra en un cromosoma no sexual. Además, la presencia de solo una de las dos copias del gen mutado es suficiente para que se desarrolle la enfermedad.
En este sentido, un individuo que posea uno de estos dos padres que padece la enfermedad tiene un 50% de riesgo de desarrollar el fenotipo enfermo él mismo.
Prevención y tratamiento.
El diagnóstico de la enfermedad es ante todo diferencial. Se basa en criterios físicos atípicos. En la mayoría de los casos, los primeros signos característicos de la enfermedad son: la presencia de ataques epilépticos recurrentes y retrasos en el desarrollo del sujeto. En otros casos, estos primeros signos dan como resultado manchas en la piel o la identificación de un tumor cardíaco.
Tras este primer diagnóstico, los exámenes adicionales son esenciales para validar el diagnóstico o no. Éstos incluyen:
- un escáner cerebral;
- una resonancia magnética (resonancia magnética) del cerebro;
- una ecografía del corazón, hígado y riñones.
El diagnóstico puede ser eficaz desde el nacimiento del niño. De lo contrario, es importante que se realice lo antes posible para poder hacerse cargo del paciente lo antes posible.
Actualmente, no existe cura para la enfermedad. Por tanto, los tratamientos asociados son independientes de los síntomas que presenta cada individuo.
Por lo general, se administran medicamentos antiepilépticos para limitar las convulsiones. Además, también se prescriben medicamentos para el tratamiento de células tumorales del cerebro y los riñones. En el contexto de los problemas de conducta, es necesario un tratamiento específico del niño.
El tratamiento de la enfermedad suele ser a largo plazo. (1)