









Contenido
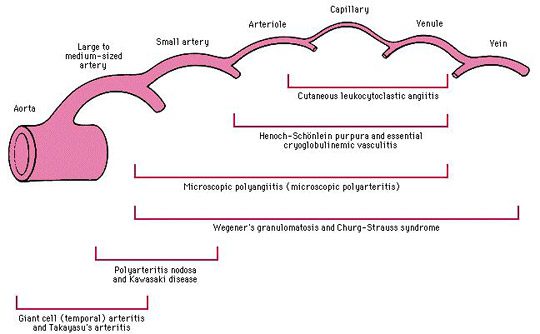
Vasculitis de pequeños capilares
Vasculitis de pequeños capilares
Se trata de un gran grupo de vasculitis de la pared de arteriolas, vénulas o capilares, cuyo pronóstico es muy variable según se trate de vasculitis cutánea pura o sistémica.
El aspecto clínico más común es la púrpura (manchas violáceas que no se desvanecen al presionarlas) abultadas e infiltradas, especialmente en los miembros inferiores, agravadas con la bipedestación, que pueden tomar varias formas (petequiales y equimóticas, necróticas, pustulosas…) o livedo, formando una especie de malla violácea (livedo reticularis) o moteada (livedo racemosa) en las piernas. También podemos observar un fenómeno de Raynaud (unos dedos se vuelven blancos con el frío).
La púrpura y el livedo pueden asociarse con otras lesiones en la piel (pápulas, nódulos, lesiones necróticas, burbujas sangrantes), urticaria fija que no pica.
La presencia de manifestaciones fuera de la piel constituye un factor de gravedad, mostrando la presencia de afectación vascular en los órganos:
- dolor en las articulaciones,
- dolor abdominal, heces negras, trastorno del tránsito,
- neuropatía periférica
- edema de las extremidades inferiores,
- Hipertensión arterial,
- dificultad para respirar, asma, tos con sangre ...
El médico prescribe exámenes destinados a buscar una causa y signos de gravedad: análisis de sangre con recuento de glóbulos, búsqueda de inflamación, pruebas de hígado y riñón, etc., búsqueda de sangre en las heces y radiografías según los puntos de llamada ( radiografía de pulmón en caso de dificultades respiratorias, etc.).
Vasculitis provocada por una infección:
- bacteriano: estreptococo, cocos gramnegativos (gonococo y meningococo)
- viral: hepatitis, mononucleosis infecciosa, VIH, etc.
- parasitaria: malaria ...
- hongos: Candida albicans ...
Vasculitis asociada a anomalías inmunológicas
- Crioglobulinemia tipo II (monoclonal mixta) y III (policlonal mixta), asociada con enfermedad autoinmune, infección (especialmente hepatitis C) o enfermedad de la sangre
- Hypocomplementémie (vascularita de urticariana de Mac Duffie)
- Hyperglobulinémie (púrpura hiperglobulinémique de Waldenström)
- Conectivitis: lupus, síndrome de Gougerot-Sjögren, artritis reumatoide ...
- Vasculitis de enfermedades de la sangre y neoplasias.
- Leucemia, linfoma, mieloma, cáncer
- Vasculitis asociada con ANCA (anticuerpos anticitoplasma de neutrófilos)
Micro Poly Angéite o MPA
La micropoliangeítis (MPA) es una angeítis necrotizante sistémica cuyos signos clínicos son muy similares a los de PAN.
El MPA está asociado con ANCA del tipo anti-mieloperoxidasa (anti-MPO) y típicamente da lugar a glomerulonefritis rápidamente progresiva y compromiso pulmonar que está ausente en PAN.
El tratamiento del MPA como del PAN comienza con la terapia con corticosteroides, a veces combinados con inmunosupresores (ciclofosfamida en particular).
Enfermedad de Wegener
La granulomatosis de Wegener es una vasculitis cuyo inicio suele estar marcado por síntomas ORL o respiratorios (sinusitis, neumopatía, etc.) resistentes a tratamientos antibióticos.
Clásicamente, la afectación difusa de ORL (pansinusitis destructiva), pulmonar (nódulos parenquimatosos) y renal (glomerulonefritis necrotizante pauciinmune creciente) produce la tríada clásica de granulomatosis de Wegener.
La membrana cutáneo-mucosa afecta aproximadamente al 50% de los pacientes: púrpura (manchas violáceas que no desaparecen al presionar) abultadas e infiltradas, pápulas, nódulos subcutáneos, ulceraciones cutáneas, pústulas, vesículas, gingivitis hiperplásica ...
ANCA es una prueba diagnóstica y evolutiva para la granulomatosis de Wegener, con fluorescencia citoplasmática difusa (c-ANCA), finamente granular con realce perinuclear y / o fluorescencia puramente perinuclear (p-ANCA).
El tratamiento de la granulomatosis de Wegener, que en ocasiones puede considerarse una urgencia médica, debe realizarse en un entorno hospitalario especializado, mediante una combinación de cortisona y ciclofosfamida oral.
Enfermedad de Churg y Strauss
El asma es un criterio mayor y precoz de esta vasculitis, que precede en promedio 8 años antes de los primeros signos de vasculitis (neuropatía, trastornos de los senos nasales, etc.) y que persiste posteriormente.
Los análisis de sangre muestran, en particular, un claro aumento de los glóbulos blancos polinucleares eosinofílicos.
El tratamiento de la enfermedad de Churg y Strauss comienza con terapia con corticosteroides, a veces combinados con inmunosupresores (especialmente ciclofosfamida).
La opinión de nuestro médico
La púrpura infiltrada (parches violáceos, algo gruesos que no desaparecen con la presión de los dedos) es el signo clave de vasculitis. Desafortunadamente, este signo no siempre está presente y la variabilidad de los signos clínicos inespecíficos a menudo dificulta el diagnóstico para los médicos. Asimismo, a menudo es difícil encontrar una causa a tratar en la vasculitis de vasos pequeños, que es con mucho el caso más importante encontrado en la práctica actual en comparación con las vasculitis de vasos medianos y grandes: aproximadamente la mitad de las vasculitis de vasos pequeños. los vasos no tienen causa encontrada durante las exploraciones biológicas y radiológicas que realiza el médico para buscar una etiología. A menudo hablamos de “vasculitis alérgica” o “vasculitis por hipersensibilidad” o más bien de “vasculitis cutánea de pequeños vasos de calibre idiopático”. Dr. Ludovic Rousseau, dermatólogo |
Puntos de interés
Grupo de estudio francés sobre vasculitis: www.vascularites.org
Dermatonet.com, sitio de información sobre piel, cabello y belleza por un dermatólogo
MedicineNet: http://www.medicinenet.com/vasculitis/article.htm