









Contenido
Enfermedad de Wilson
Qué es ?
La enfermedad de Wilson es una enfermedad genética hereditaria que impide la eliminación del cobre del organismo. La acumulación de cobre en el hígado y el cerebro provoca problemas hepáticos o neurológicos. La prevalencia de la enfermedad de Wilson es muy baja, alrededor de 1 de cada 30 personas. (000) Existe un tratamiento eficaz para esta enfermedad, pero su diagnóstico precoz es problemático porque permanece en silencio durante mucho tiempo.
Síntomas
La acumulación de cobre comienza al nacer, pero los primeros síntomas de la enfermedad de Wilson a menudo no aparecen hasta la adolescencia o la edad adulta. Pueden ser muy diversos porque varios órganos se ven afectados por la acumulación de cobre: el corazón, los riñones, los ojos, la sangre… Los primeros signos son hepáticos o neurológicos en tres cuartas partes de los casos (40% y 35% respectivamente), pero pueden también serán psiquiátricos, renales, hematológicos y endocrinológicos. El hígado y el cerebro se ven particularmente afectados porque ya contienen la mayor cantidad de cobre de forma natural. (2)
- Trastornos hepáticos: ictericia, cirrosis, insuficiencia hepática ...
- Trastornos neurológicos: depresión, trastornos del comportamiento, dificultades de aprendizaje, dificultades para expresarse, temblores, calambres y contracturas (distonía) ...
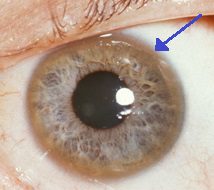
El anillo Keyser-Fleisher que rodea el iris es característico de la acumulación de cobre en el ojo. Además de estos síntomas agudos, la enfermedad de Wilson puede presentarse con síntomas inusuales, como fatiga general, dolor abdominal, vómitos y pérdida de peso, anemia y dolor articular.
Los orígenes de la enfermedad
En el origen de la enfermedad de Wilson, existe una mutación en el gen ATP7B ubicado en el cromosoma 13, que participa en el metabolismo del cobre. Controla la producción de una proteína ATPasa 2 que desempeña un papel en el transporte de cobre desde el hígado a otras partes del cuerpo. El cobre es un componente necesario para muchas funciones celulares, pero en exceso se vuelve tóxico y daña tejidos y órganos.
Los factores de riesgo
La transmisión de la enfermedad de Wilson es autosómica recesiva. Por tanto, es necesario recibir dos copias del gen mutado (del padre y de la madre) para desarrollar la enfermedad. Esto significa que los hombres y las mujeres están igualmente expuestos y que dos padres que portan el gen mutado pero que no están enfermos tienen un riesgo en cuatro en cada nacimiento de transmitir la enfermedad.
Prevención y tratamiento.
Existe un tratamiento eficaz para detener la progresión de la enfermedad y reducir o incluso eliminar sus síntomas. También es necesario que se inicie de forma precoz, pero a menudo se necesitan muchos meses después del inicio de los síntomas para diagnosticar esta enfermedad silenciosa, poco conocida y cuyos síntomas apuntan a muchas otras afecciones (hepatitis para la que hay daño hepático y depresión por afectación psiquiátrica). .
Un tratamiento “quelante” permite atraer el cobre y eliminarlo en la orina, limitando así su acumulación en los órganos. Se basa en D-penicilamina o trientina, medicamentos que se toman por vía oral. Son eficaces, pero pueden provocar efectos secundarios graves (daño renal, reacciones alérgicas, etc.). Cuando estos efectos secundarios son demasiado importantes, recurrimos a la administración de zinc que limitará la absorción de cobre por los intestinos.
Puede ser necesario un trasplante de hígado cuando el hígado está demasiado dañado, que es el caso del 5% de las personas con enfermedad de Wilson (1).
Se ofrece una prueba de detección genética a los hermanos de una persona afectada. Da lugar a un tratamiento preventivo eficaz en el caso de que se detecte una anomalía genética en el gen ATP7B.