









Contenido
Síndrome de Treacher-Collins
Una enfermedad genética rara, el síndrome de Teacher-Collins se caracteriza por el desarrollo de defectos congénitos del cráneo y la cara durante la vida embrionaria, lo que resulta en deformidades de la cara, los oídos y los ojos. Las consecuencias estéticas y funcionales son más o menos graves y algunos casos requieren numerosas intervenciones quirúrgicas. Sin embargo, en la mayoría de los casos, hacerse cargo permite preservar una cierta calidad de vida.
¿Qué es el síndrome de Treacher-Collins?
Definición
El síndrome de Treacher-Collins (llamado así por Edward Treacher Collins, quien lo describió por primera vez en 1900) es una rara enfermedad congénita que se manifiesta desde el nacimiento con malformaciones más o menos graves de la parte inferior del cuerpo. cara, ojos y oídos. Los ataques son bilaterales y simétricos.
Este síndrome también se denomina síndrome de Franceschetti-Klein o disostosis mandibulo-facial sin anomalías finales.
Causas
Hasta ahora se sabe que tres genes están involucrados en este síndrome:
- el gen TCOF1, ubicado en el cromosoma 5,
- los genes POLR1C y POLR1D, ubicados en los cromosomas 6 y 13 respectivamente.
Estos genes dirigen la producción de proteínas que juegan un papel clave en el desarrollo embrionario de las estructuras faciales. Su alteración por mutaciones altera el desarrollo de las estructuras óseas (principalmente las de la mandíbula superior e inferior y los pómulos) y los tejidos blandos (músculos y piel) de la parte inferior del rostro durante el segundo mes de gestación. El pabellón auricular, el canal auditivo y las estructuras del oído medio (huesecillos y / o tímpanos) también se ven afectados.
Diagnóstico
Se pueden sospechar malformaciones faciales a partir de la ecografía del segundo trimestre del embarazo, especialmente en casos de malformaciones importantes del oído. En este caso, el diagnóstico prenatal será establecido por un equipo multidisciplinar a partir de la resonancia magnética (RM) del feto, permitiendo visualizar las malformaciones con mayor precisión.
La mayoría de las veces, el diagnóstico se realiza mediante un examen físico que se realiza al nacer o poco después. Debido a la gran variabilidad de malformaciones, debe confirmarse en un centro especializado. Es posible que se solicite una prueba genética en una muestra de sangre para buscar las anomalías genéticas involucradas.
Algunas formas leves pasan desapercibidas o se detectarán fortuitamente tarde, por ejemplo, tras la aparición de un nuevo caso en la familia.
Una vez que se hace el diagnóstico, el niño es sometido a una serie de exámenes adicionales:
- imágenes faciales (rayos X, tomografía computarizada y resonancia magnética),
- exámenes de oído y pruebas de audición,
- evaluación de la visión,
- buscar apnea del sueño (polisomnografía)…
La gente interesada
Se cree que el síndrome de Treacher-Collins afecta a uno de cada 50 recién nacidos, tanto niñas como niños. Se estima que en Francia aparecen cada año alrededor de 000 casos nuevos.
Los factores de riesgo
Se recomienda la asesoría genética en un centro de referencia para evaluar los riesgos de transmisión genética.
Alrededor del 60% de los casos aparecen de forma aislada: el niño es el primer paciente de la familia. Las malformaciones se producen a raíz de un accidente genético que afectó a una u otra de las células reproductoras implicadas en la fecundación (mutación “de novo”). El gen mutado luego se transmitirá a sus descendientes, pero no existe un riesgo particular para sus hermanos. Sin embargo, debe comprobarse si uno de sus padres no padece realmente una forma menor del síndrome y es portador de la mutación sin saberlo.
En otros casos, la enfermedad es hereditaria. La mayoría de las veces, el riesgo de transmisión es uno de cada dos con cada embarazo, pero dependiendo de las mutaciones involucradas, existen otras formas de transmisión.
Síntomas del síndrome de Treacher-Collins
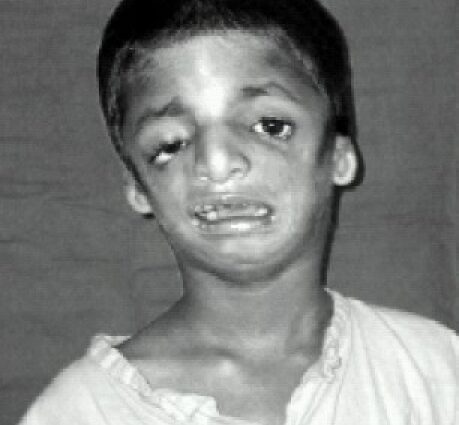
Los rasgos faciales de los afectados suelen ser característicos, con mentón atrofiado y retraído, pómulos inexistentes, ojos inclinados hacia las sienes, orejas con pabellón pequeño y mal dobladillo, o incluso completamente ausentes ...
Los principales síntomas están relacionados con malformaciones de la esfera ORL:
Dificultades respiratorias
Muchos niños nacen con vías respiratorias superiores estrechas y bocas muy abiertas, con una pequeña cavidad bucal obstruida en gran parte por la lengua. De ahí las importantes dificultades respiratorias, especialmente en recién nacidos y lactantes, que se expresan por ronquidos, apnea del sueño y respiración demasiado débil.
Dificultad para comer
En los bebés, la lactancia materna puede verse obstaculizada por la dificultad para respirar y por anomalías del paladar y el paladar blando, a veces dividido. La alimentación es más fácil después de la introducción de alimentos sólidos, pero masticar puede ser difícil y los problemas dentales son comunes.
Sordera
El malestar auditivo debido a malformaciones del oído externo o medio está presente en 30 a 50% de los casos.
Alteraciones visuales
Un tercio de los niños padece estrabismo. Algunos también pueden ser miopes, hipermétropes o astigmáticos.
Dificultades de aprendizaje y comunicación.
El síndrome de Treacher-Collins no causa un déficit intelectual, pero la sordera, los problemas visuales, las dificultades del habla, las repercusiones psicológicas de la enfermedad y las alteraciones inducidas por una atención médica a menudo muy intensa pueden causar un retraso. lenguaje y dificultad para comunicarse.
Tratamientos para el síndrome de Treacher-Collins
Cuidado infantil
Puede ser necesario un soporte respiratorio y / o alimentación por sonda para facilitar la respiración y la alimentación del bebé, a veces desde el nacimiento. Cuando la asistencia respiratoria debe mantenerse en el tiempo, se realiza una traqueotomía (pequeña abertura en la tráquea, en el cuello) para introducir una cánula que directamente asegura el paso del aire en las vías respiratorias.
Tratamiento quirúrgico de malformaciones.
Se pueden proponer intervenciones quirúrgicas más o menos complejas y numerosas, relativas al paladar blando, mandíbula, mentón, orejas, párpados y nariz para facilitar la alimentación, la respiración o la audición, pero también para reducir el impacto estético de las malformaciones.
Como indicación, las hendiduras del paladar blando se cierran antes de los 6 meses de edad, los primeros procedimientos estéticos en los párpados y pómulos a partir de los 2 años, el alargamiento de la mandíbula (distracción mandibular) hacia los 6 o 7 años, la reinserción de el pabellón auricular alrededor de los 8 años, agrandamiento de los conductos auditivos y / o cirugía de los huesecillos alrededor de los 10 a 12 años ... Otras operaciones de cirugía estética aún se pueden realizar en la adolescencia ...
Audífono
Los audífonos a veces son posibles a partir de los 3 o 4 meses cuando la sordera afecta a ambos oídos. Se dispone de diferentes tipos de prótesis según la naturaleza del daño, con buena eficacia.
Seguimiento médico y paramédico
Para limitar y prevenir la discapacidad, el seguimiento periódico es multidisciplinar y requiere de varios especialistas:
- ENT (alto riesgo de infección)
- Oftalmólogo (corrección de alteraciones visuales) y ortoptista (rehabilitación ocular)
- Dentista y ortodoncista
- Logopeda…
A menudo se necesita apoyo psicológico y educativo.